Question #706c0
1 Answer

I'll help with problems 1 and 2, but not 3, because it would make this too long.
Here are the main points:
- The MO diagram can be found here.
- Purely from the angular overlap method perspective, square planar is favored because there is
1.33e_(sigma) lesssigma destabilization.
DISCLAIMER: LONG ANSWER!
POINT GROUP AND SYMMETRY ELEMENTS
For an
 https://upload.wikimedia.org/
https://upload.wikimedia.org/
You should work through this and find the symmetry elements belonging to the
(NOTE: You only need to identify
hatE , the identity, because everything has it.hatC_4^n(z) , the principal axis of 4-fold rotation symmetry. You can use this up to3 times before getting the identity back.hatC_2' , an axis of 2-fold rotation symmetry on thexy plane, along a trans"B"-"A"-"B" bond.hatC_2'' , a rotation axis of 2-fold symmetry on thexy plane, bisecting a cis"B"-"A"-"B" bond.hatsigma_v , a vertical mirror plane colinear with thehatC_2' axis, along a trans"B"-"A"-"B" bond.hatsigma_d , a dihedral mirror plane colinear with thehatC_2'' axis, bisecting a cis"B"-"A"-"B" bond.hatsigma_h , a horizontal mirror plane on thexy plane.hatS_4 , an improper rotation axis of 4-fold symmetry, becausehatS_4 = hatC_4hatsigma_h , which must be in the point group from the properties of a group (any element in a point group can be generated by multiplication of two other elements in the group).hati , an inversion center, because all the"B" ligands are identical and there are an even number of them. Thus,(x,y,z) = (-x,-y,-z) for each of them.
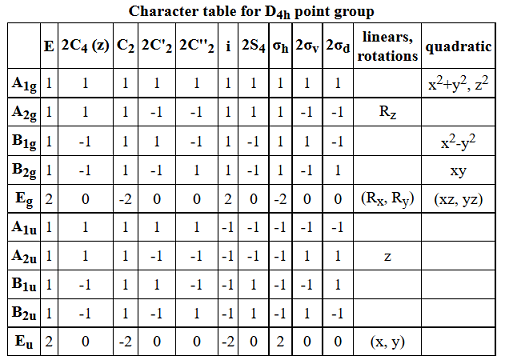
CHARACTER TABLE
Its character table, which you should have in front of you, is:
 https://www.webqc.org/
https://www.webqc.org/
I assume you know a few features of the character table, such as:
- The sum of the coefficients of the rotation operators
hatR gives the orderh of the group. - The
A//B andE irreducible representations (IRREPs) are one-fold and two-fold degenerate, respectively. This is why the character ofE_g underhatE is2 and not1 . - The "linear" column gives you the orbitals that transform under particular symmetries (
p_x,p_y,p_z ). - The "quadratic" column gives you the orbitals that transform under those particular symmetries (
overbrace(s)^(x^2 + y^2), d_(z^2), d_(x^2-y^2), d_(xy), [d_(xz),d_(yz)] ).
The next thing to do is to generate the reducible representation for the ligand orbitals. Without doing that, we won't know which metal orbitals match.
Since we want only
However, when doing this for
GENERATING THE REDUCIBLE REPRESENTATION:
The reducible representation
- If the operation returns the orbital unmoved, put
bb1 in the reducible representation. - If the operation returns the orbital with the opposite phase, put
bb(-1) in the reducible representation. - If the operation returns the orbital moved from where it was before, put
bb0 in the reducible representation.
The results are:
" "" "hatE" "hatC_4" "hatC_2" "hatC_2'" "hatC_2''" "hati" "hatS_4" "hatsigma_h" "hatsigma_v" "hatsigma_d
Gamma_s = 4" "0" "" "0" "color(white)(.)2" "" "0" "" "color(white)(.)0" "0" "color(white)(.)4" "color(white)(.)2" "color(white)(.)0
" "color(white)(.,.)hatE" "hatC_4" "hatC_2" "hatC_2'" "hatC_2''" "hati" "hatS_4" "hatsigma_h" "hatsigma_v" "hatsigma_d
Gamma_(p_y) = 4" "0" "" "0" "color(white)(.)2" "" "0" "" "color(white)(.)0" "0" "color(white)(.)4" "color(white)(.)2" "color(white)(.)0
REDUCING TO A SET OF IRREPS:
Here we seek two or more IRREPs whose line of characters adds up to this. Among them must be the totally symmetric one,
Gamma_s - Gamma_(A_(1g))
= 3" "-1" "-1" "1" "-1" "-1" "-1" "3" "1" "-1
With an even number of orbitals, you can choose their phase so that trans ligands have the opposite phase and cis ligands have same phase. This is antisymmetric with respect to inversion, so
Gamma_s - Gamma_(A_(1g)) - Gamma_(E_u)
= 1" "-1" "1" "1" "-1" "1" "-1" "1" "1" "-1
And by inspection of the character table, this row of characters matches with
color(blue)(Gamma_s = A_(1g) + B_(1g) + E_u)
color(blue)(Gamma_(p_y) = A_(1g) + B_(1g) + E_u)
METAL ORBITAL SYMMETRIES
This isn't as complicated. You can look at the character table and directly read these off to be:
" "d_(z^2) " "harr A_(1g)
" "d_(x^2-y^2) harr B_(1g)
" "color(red)(d_(xy)) " "harr color(red)(B_(2g)) (nonbonding)
[color(red)(d_(xz), d_(yz))] harr color(red)(E_u) (nonbonding)
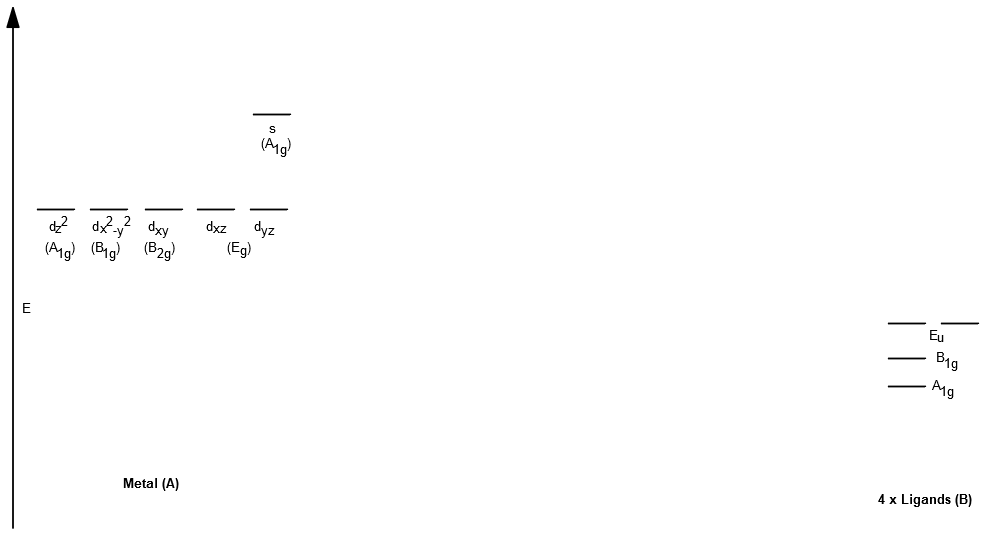
The orbitals with different symmetries don't interact. So, we get the following interactions:
"Metal" s withA_(1g) , making ana_(1g) bonding anda_(1g)^"*" antibonding MO.Although
d_(z^2) isA_(1g) , it is relatively nonbonding because there are no ligands on thez axis. However, due to the metals orbital and the ligandA_(1g) orbitals, there is some stabilization even without direct interaction.
"Metal" d_(x^2-y^2) with ligandB_(1g) , making ab_(1g) bonding andb_(1g)^"*" antibonding MO.
"Metal" color(red)(d_(xy)) (color(red)(B_(2g)) ) orbital becomes EXACTLY nonbonding due to no matching orbital symmetries.
"Metal" color(red)(d_(xz), d_(yz)) (color(red)(E_u) ) orbitals become EXACTLY nonbonding (ignoring metalp orbitals).
This results in the following orbital diagram without the MOs so far (ignoring metal
[
When you make the MOs, use relative energy orderings and you should get something like this:
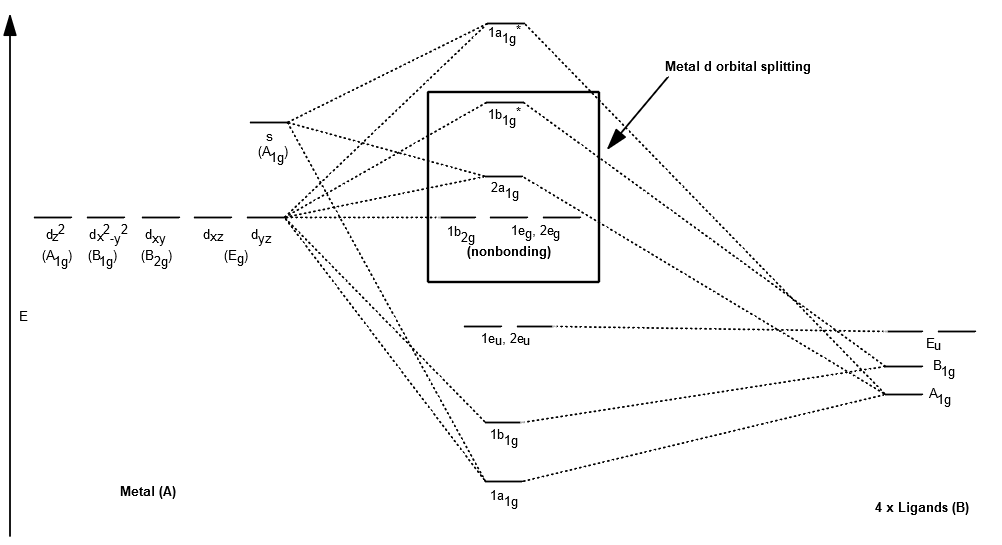
Note that this won't exactly match the full square planar d orbital splitting diagram because we neglected the
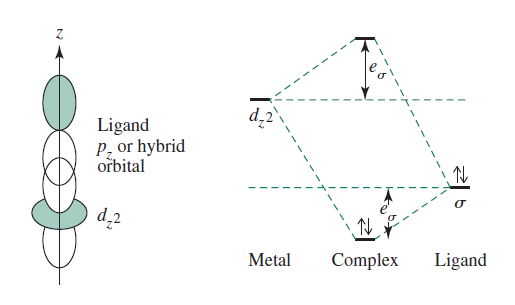
ANGULAR OVERLAP METHOD
For
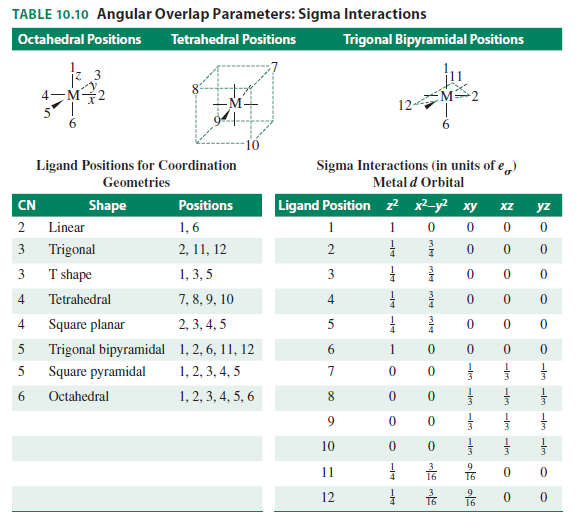 Inorganic Chemistry, Miessler et al., pg. 384
Inorganic Chemistry, Miessler et al., pg. 384
- For square planar, ignore positions
1 and6 in the octahedral diagram. - For tetrahedral, use the central diagram.
Since we only consider
 Inorganic Chemistry, Miessler et al., pg. 383
Inorganic Chemistry, Miessler et al., pg. 383
From the table for square planar,
d_(z^2) is destabilized by1/4e_sigma due to ligands2,3,4,5 (rows 2 - 5, column 2). This adds up tocolor(blue)(e_sigma) .-
d_(x^2-y^2) is destabilized by3/4e_sigma due to ligands2,3,4,5 (rows 2 - 5, column 3). This adds up tocolor(blue)(3e_sigma) . -
The
xy ,xz , andyz are nonbonding because they have no destabilizing or stabilizing contribution (rows 3 - 5, columns 4 - 6).
From the table for tetrahedral,
-
d_(xy) ,d_(xz) , andd_(yz) are all destabilized by1/3e_sigma due to ligands7,8,9,10 (rows 7 - 10, columns 4 - 6). This adds up tocolor(blue)(4/3e_sigma) for each orbital. -
The
z^2 andx^2-y^2 are nonbonding because they have no destabilizing or stabilizing contribution (rows 7 - 10, columns 2 - 3).
Based purely on the angular overlap method, since the ligands are destabilizing the metal
e_sigma + 3e_sigma = color(blue)(4e_(sigma)) in a square planar regime
and
4 xx 4/3e_sigma = color(blue)(5.33e_(sigma)) in a tetrahedral regime,
the square planar shape is energetically favored. This is an OK approximation because the
