Another Thermo Question?
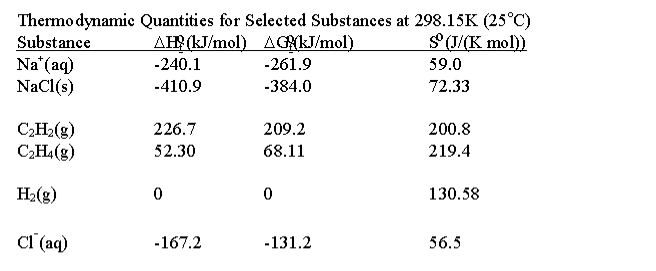
Consider the reaction
C2H2(g) + H2(g) <->C2H4(g). Use the data in
the table above for the following:
A: Estimate Delta G, H and S for the reaction above at 25C, and
use your results to show that the reaction is spontaneous at room
temperature.
B: As the temperature increases, the reaction ceases to be
spontaneous. Use the Gibbs-Helmholtz equation to predict the
temperature at which the reaction is no longer spontaneous for 1 atm
pressure in each gas.
Consider the reaction
C2H2(g) + H2(g) <->C2H4(g). Use the data in
the table above for the following:
A: Estimate Delta G, H and S for the reaction above at 25C, and
use your results to show that the reaction is spontaneous at room
temperature.
B: As the temperature increases, the reaction ceases to be
spontaneous. Use the Gibbs-Helmholtz equation to predict the
temperature at which the reaction is no longer spontaneous for 1 atm
pressure in each gas.
1 Answer

PART A
Part A is fairly straightforward, and is just asking you to choose the thermodynamic values to use. Let
If
#DeltaY_"rxn"^@ = sum_P nu_P DeltaY_(f,P)^@ - sum_R nu_R DeltaY_(f,R)^@#
If
#DeltaS_"rxn"^@ = sum_P nu_P S_(P)^@ - sum_R nu_R S_(R)^@# where:
#DeltaY_f^@# is some thermodynamic function of formation (such as Gibbs energy of formation or enthalpy of formation) for one pure substance.#S^@# is the standard molar entropy for one pure substance, and#P# (or#R# ) stands for products (or reactants). This is defined a little differently because the initial state is at#"0 K"# , and#S_("0 K") = 0# from the third law of thermodynamics.#nu# is the stoichiometric coefficient of the substance in the balanced chemical reaction.
#"C"_2"H"_2(g) + "H"_2(g) rightleftharpoons "C"_2"H"_4(g)#
We'll choose
#color(blue)(DeltaG_"rxn"^@) = nu_(C_2H_4(g))DeltaG_(f,C_2H_4(g))^@ - [nu_(C_2H_2(g))DeltaG_(f,C_2H_2(g))^@ + nu_(H_2(g))DeltaG_(f,H_2(g))^@]#
#= (1)("68.11 kJ/mol") - [(1)("209.2 kJ/mol") +(1) ("0 kJ/mol")]#
#= color(blue)(-"141.1 kJ/mol")#
Already we can say that the reaction is spontaneous at room temperature, since
Essentially the same math follows for
#color(blue)(DeltaH_"rxn"^@) = nu_(C_2H_4(g))DeltaH_(f,C_2H_4(g))^@ - [nu_(C_2H_2(g))DeltaH_(f,C_2H_2(g))^@ + nu_(H_2(g))DeltaH_(f,H_2(g))^@]#
#= (1)("52.30 kJ/mol") - [(1)("226.7 kJ/mol") +(1) ("0 kJ/mol")]#
#= color(blue)(-"174.4 kJ/mol")#
which says the reaction is exothermic, i.e. it releases energy into the surroundings.
And for
#DeltaG_"rxn"^@ = DeltaH_"rxn"^@ - TDeltaS_"rxn"^@#
and solve for
#color(blue)(DeltaS_"rxn"^@) = (DeltaH_"rxn"^@ - DeltaG_"rxn"^@)/T#
#= color(blue)(-"111.69 J/mol"cdot"K")#
(don't forget to convert from#"kJ"# to#"J"# ).
which means the entropy of the system decreases, meaning that the entropy of the surroundings increases.
Either way, there's about a
PART B
I'm surprised you are asked to use the Gibbs-Helmholtz equation... Because this is what I see on Wikipedia:
#((del(DeltaG_("rxn")^@"/"T))/(delT))_P = -(DeltaH_("rxn")^@)/T^2#
If we want the reaction to no longer be spontaneous, then we must have that
The easy way
#cancel(DeltaG_"rxn"^@)^"assumed 0" = DeltaH_"rxn" - T_"nonspont."DeltaS_"rxn"^@#
#=> DeltaH_"rxn"^@ = T_"nonspont."DeltaS_"rxn"^@#
#color(blue)(T_"nonspont.") = (DeltaH_"rxn"^@)/(DeltaS_"rxn"^@)#
#= (-"174.4 kJ/mol")/(-"0.11169 kJ/mol")#
#=# #color(blue)("1561.5 K")#
The hard way
From
Let us multiply both sides by
#d(DeltaG_"rxn"^@"/"T) = -(DeltaH_"rxn"^@)/T^2dT#
Integrating both sides from the initial temperature
#int_(T^@)^(T_"nonspont.") d(DeltaG_"rxn"^@"/"T') = DeltaH_"rxn"^@int_(T^@)^(T_"nonspont.")-1/(T')^2dT'#
where we have assumed that
The integral of a derivative cancels out on the left side, and the right side integrates to give
#|[(DeltaG_"rxn"^@(T'))/(T')]|_(T^@)^(T_"nonspont.") = DeltaH_"rxn"^@|[(1/(T'))]|_(T^@)^(T_"nonspont.")#
#(DeltaG_"rxn"^@(T_"nonspont."))/T_"nonspont." - (DeltaG_"rxn"^@(T^@))/T^@ = DeltaH_"rxn"^@[1/(T_"nonspont.") - 1/T^@]#
Now, if we set
#cancel((DeltaG_"rxn"^@(T_"nonspont."))/T_"nonspont.")^(0) - (stackrel("Known")overbrace(DeltaG_"rxn"^@(T^@)))/T^@ = stackrel("Known")overbrace(DeltaH_"rxn"^@)[1/T_"nonspont." - 1/T^@]#
Now we'd solve for
#-(DeltaG_"rxn"^@(T^@))/(DeltaH_"rxn"^@T^@) = 1/T_"nonspont." - 1/T^@#
#1/T^@ - (DeltaG_"rxn"^@(T^@))/(DeltaH_"rxn"^@T^@) = 1/T_"nonspont."#
#=> color(blue)(T_"nonspont.") = [1/T^@ - (DeltaG_"rxn"^@(T^@))/(DeltaH_"rxn"^@T^@)]^(-1) = (1/T^@)^(-1)[1 - (DeltaG_"rxn"^@(T^@))/(DeltaH_"rxn"^@)]^(-1)#
#= T^@[1 - (DeltaG_"rxn"^@(T^@))/(DeltaH_"rxn"^@)]^(-1) = ("298.15 K")[1 - (-"141.1 kJ/mol")/(-"174.4 kJ/mol")]^(-1)#
#=# #color(blue)("1561.5 K")#
and you can see the two approaches essentially give the same result (
