Steric Strain
Key Questions
-
Steric strain is most evident in cyclic molecules because the ring structure prevents the groups from getting away from each other.
Steric strain is the increase in potential energy of a molecule due to repulsion between groups on non-neighbouring carbons.
Consider a non-cyclic molecule such as 1,3-dichloropropane. We could draw it as
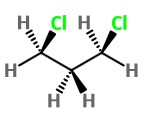
Here, the electron clouds of the Cl atoms are close together, and they repel each other. But there is free rotation about the C-C σ bonds. The CH₂Cl groups can rotate to get the Cl atoms further apart.
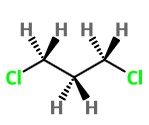
This rotation is severely hindered when the C atoms are in a ring.
For example, a cyclohexane ring is most stable in a chair conformation.
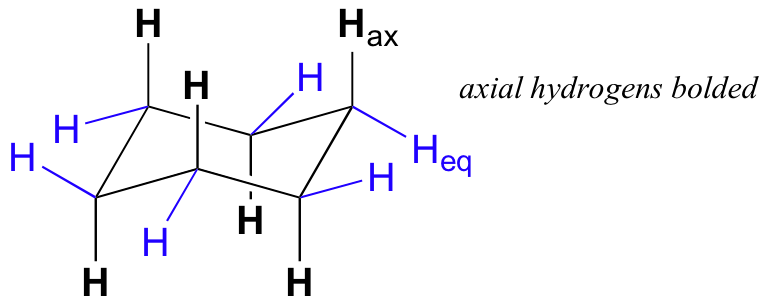
If you had a cyclohexane with two groups in axial positions, you would get steric strain because these groups would be too close and they would repel each other.
The molecule below has steric strain from many 1,3-diaxial interactions.

Most of these can be relieved by flipping the cyclohexane chair. But then the Br would be axial and have its own 1,3-diaxial steric repulsions.
 Ernest Z.
·
1
·
Jan 31 2015
Ernest Z.
·
1
·
Jan 31 2015
-
Steric strain is the increase in potential energy of a molecule that is caused by van der Waals repulsions and cannot be reduced by rotation around a single bond.
We often find steric strain in alkenes and ring systems.
Alkenes

van der Waals repulsions between the eclipsed chlorine atoms cannot be reduced by rotation around the rigid carbon-carbon bond.
This is why cis alkenes are less stable than trans alkenes.
Ring systems
Groups in the axial positions of cyclohexane experience van der Waals repulsions from the other axial groups on the same side of the ring.

No such problems exist for equatorial substituents
This is why equatorial positions are more stable than axial ones.
Ernest Z.
·
1
·
May 20 2015