How would you distinguish between ortho and para nitrophenol using infrared spectroscopy?
1 Answer

The thing is, anyone would have a hard time justifying which of these two compounds they had made from using just an IR spectrum, even if they had both.
STRUCTURES AND PREDICTED VIBRATIONAL MOTIONS
The structures are:
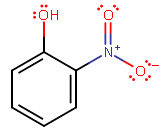 (o-nitrophenol)
(o-nitrophenol)
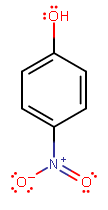 (p-nitrophenol)
(p-nitrophenol)
The peaks you should see based on the structure of the molecule (and based on what I have in my reference text, Techniques in Organic Chemistry, Mohrig) are:
\mathbf("C"="C") aromatic (stretch):1620~1440 "cm"^(-1) ; medium to weak; sharp\mathbf("C"-"H") aromatic (stretch, bend):3100~3000, 900~680 "cm"^(-1) , respectively; medium to weak; sharp\mathbf("C"-"O") alcohol (stretch):1300~1000 "cm"^(-1) ; strong; sharp\mathbf("O"-"H") alcohol (stretch):3650~3200 "cm"^(-1) ; medium to strong; broad\mathbf("NO"_2) nitro (stretch):1570~1490, 1390~1300 "cm"^(-1) ; strong; sharp
THE IR SPECTRA
Here are two sample spectra run on a KBr pellet (I seem to recall them being a few thousand dollars each?):
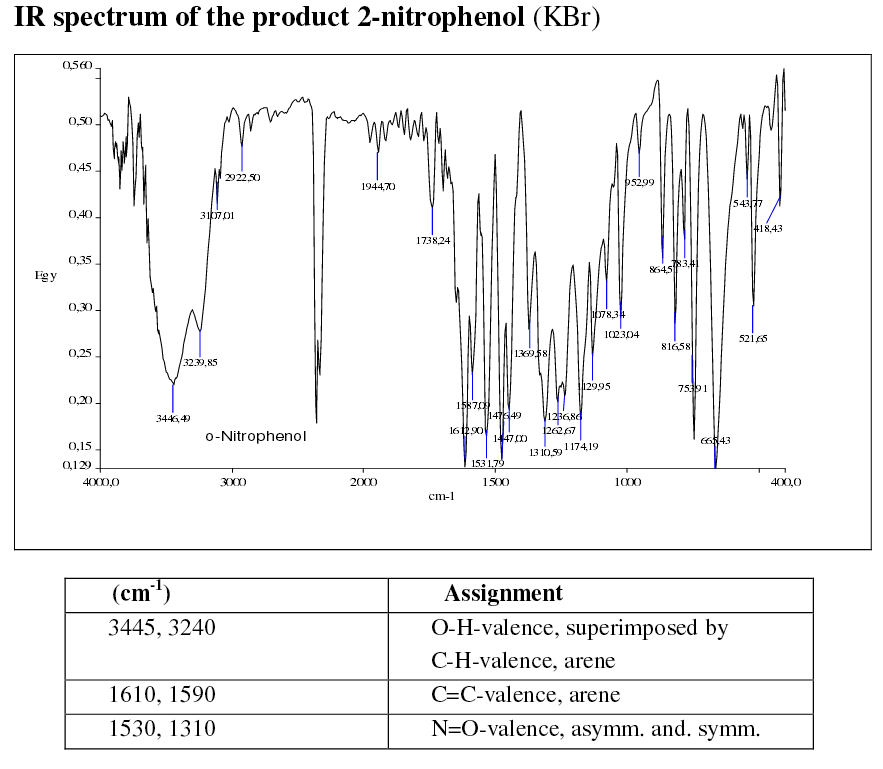 http://www.oc-praktikum.de/
http://www.oc-praktikum.de/
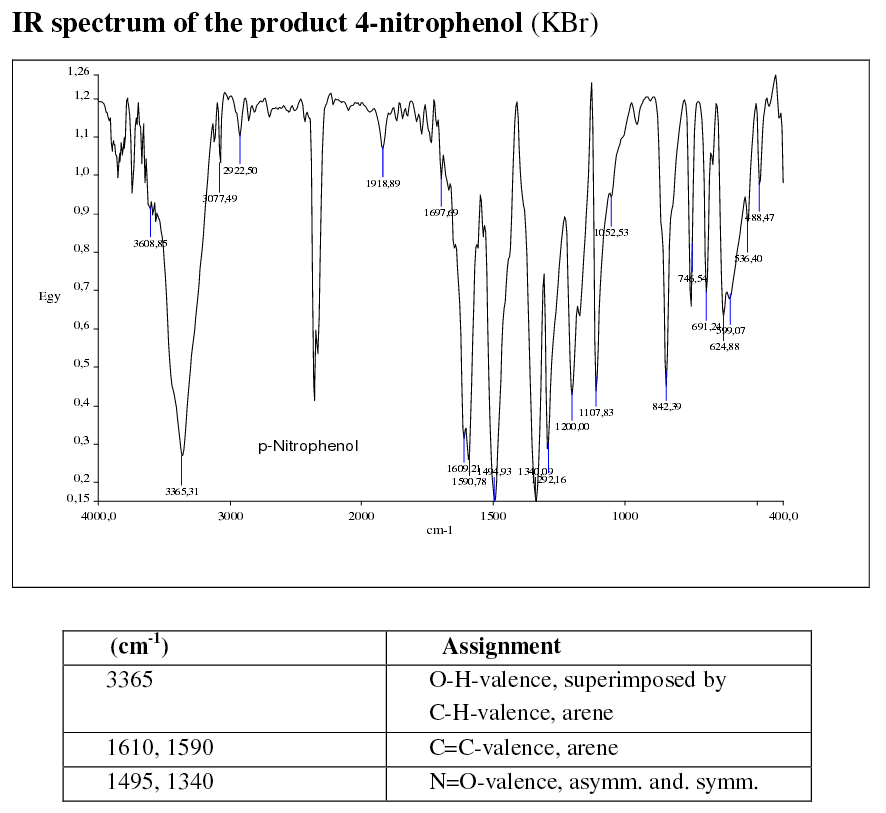 http://www.oc-praktikum.de/
http://www.oc-praktikum.de/
You should notice that the spectra are very similar above
Also, you may notice though is that the ortho spectrum is messier below
That's important; why are they stronger for the ortho isomer?
PEAKS IN IR SPECTRA ARISE FROM A CHANGE IN DIPOLE MOMENT
One thing you should remember is that peaks in IR spectra arise from a change in dipole moment during the sample analysis. You can probably imagine that changes in dipole moment are lessened when there is more symmetry in the molecule.
Not surprisingly, the main difference between the two compounds is thus the amount of symmetry they have---notice how the para isomer has a (
MORE SYMMETRY REDUCES THE CHANGE IN DIPOLE MOMENT
The vector direction of the nitro group stretches on the para isomer is more symmetrical with respect to the vector direction of the hydroxyl group stretches than in the ortho isomer, which reduces the overall change in dipole moment during those stretches, thus weakening any peaks pertaining to those vibrational motions that are predicted to show up in the IR spectrum.
As a result, we see weaker peaks in the more symmetrical para isomer for the
ALTERNATIVE APPROACH?
Of course, had you not had BOTH spectra in front of you, you might not have been able to notice such subtleties. So, I wouldn't say this is a practical way to approach the determination of which product you actually made more of during your synthesis.
You should also consider your
