NMR to Molecule
Key Questions
-
Answer:
Yes Absolutely
Explanation:
The technique DEPT gives similar information to an 'Off-resonance decoupled spectra' (one bond
#C-H# couplings are retained so the single for a particular#C# is given by the number of#H# bonded,#n+1# rule, eg. for a#-CH2-# we would see a triplet).
Without going to deep into the physics DEPT is performed by a sequence of pulses with different delay times to create a DEPT spectra. This would result in identification of#-CH_3# and#CH# , these would appear as a normal peak pointing upwards. And the#-CH_2-# group would appear as an inverted peak.
This would make it possible to figure out how many H there are bonded to C.
So DEPT would often be performed together with an 'Off-resonance decoupled spectra', making it easy to detect the different#C-H# groups. Shulgin
·
1
·
Mar 31 2018
Shulgin
·
1
·
Mar 31 2018
-
This is more of a "how to circumvent complicated spectra" answer, but has some stuff on dealing with them.
Sometimes it's because of your shimming. Or maybe the compound is just too complicated. Or, maybe it's because you didn't normalize the peaks. Or you have a solvent with clashing peaks.
Oftentimes, complex spectra either should not be analyzed (e.g. complex protein) or should be refined before analysis (e.g. poor NMR workup).
Whatever the case, to minimize the complications:
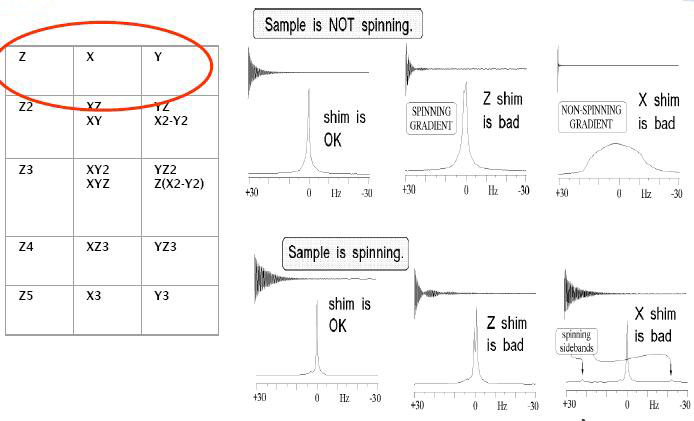
- Work on shimming more precisely to make sure proton-splitting peaks are as even as possible.
This ensures that you can say with good certainty that the relative peak heights you see indeed correspond to proton splitting, and not two nearby but coincidentally unrelated peaks.
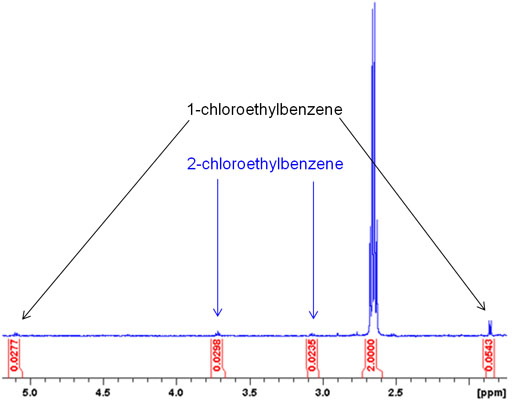
- Separate/extract contaminants from your solution if possible to clean up extraneous peaks. Recognize which peaks might just be leftover reactant.
This minimizes the possibility of misidentifying peaks that might overlap with your more important peaks, making them seem stronger.
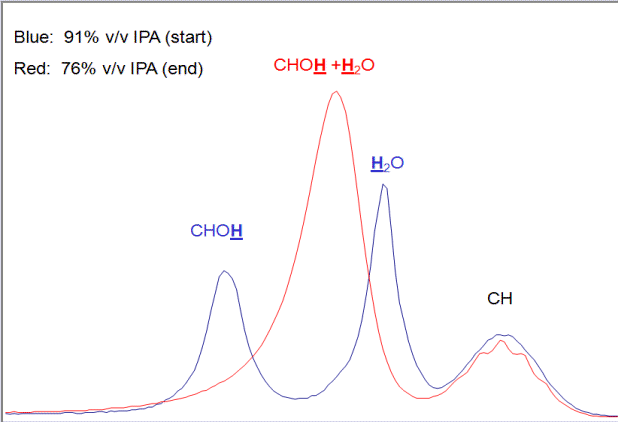
- Use solvents that have well-known, distinguishable peaks that don't coincide with many analyte peaks.
You should be able to identify where the solvent is at all times if possible. If you can't, there's a decent chance it is interfering with other peaks' heights and widths.
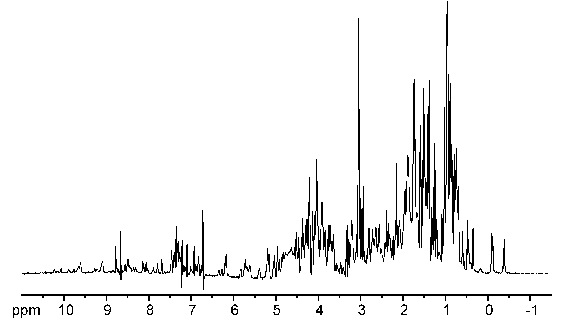
- Clump together certain peaks rather than being as specific as possible. If you don't know what each individual peak is, and you have a good reason why, I see no reason why you should identify which peaks are precisely what.
- Some good reasons:
- "too much peak overlap" (e.g. this is often true with aromatic compounds. You will likely see a bunch of aromatic peaks near
#"7~9 ppm"# , and if you do, for a great deal of cases, it's fine to lump all the aromatic protons together and write, "aromatic protons on bottom left ring" or similar.) - "peaks are too weak to tell whether or not they are actual peaks" (e.g. very slight contaminants or leftover reactants)
- "too unresolved" (e.g. a doublet looking like a singlet with a tiny notch at the top)
- "The compound is a complex protein" (e.g. too many amino acids giving rise to unresolvable peaks)
- "too much peak overlap" (e.g. this is often true with aromatic compounds. You will likely see a bunch of aromatic peaks near
- Some good reasons:
 Truong-Son N.
·
1
·
Sep 29 2015
Truong-Son N.
·
1
·
Sep 29 2015
- Work on shimming more precisely to make sure proton-splitting peaks are as even as possible.